Phenylketonuria is considered a rather rare medical condition, to be more precise this is an infrequent genetic condition that develops due to the presence of a defective gene, the one in charge of metabolizing the amino acid phenylalanine.
Rare Inherited Condition
The condition is inherited and runs in certain families. The defective gene is transferred in an autosomal recessive fashion, meaning that both parents must possess the defective gene.
The affected gene makes a person incapable of metabolizing the amino acid phenylalanine. This acid is a part of food containing proteins. If such food is introduced to the body, phenylalanine will not be metabolized but instead will gradually accumulate in the blood and eventually trigger certain problems. The build-up of phenylalanine is most detrimental for the brain and the central nervous system in general. Therefore, the condition must be diagnosed on time and treated adequately. Only with proper treatment, patients will not suffer permanent neurological and brain damage.
When it comes to symptoms and signs of phenylketonuria, these typically include head size significantly below normal, delayed mental and social skills, mental retardation, hyperactivity, jerky movements of the extremities (seizures), tremor and unusual positioning of the hands. Since phenylalanine plays important role in production of melanin, the pigment normally found in the skin, hair, mucous membranes and several more body organs, these patients have lighter skin, hair and eyes compared to their siblings not affected by the disease. Excess of phenylalanine is also blamed for skin rash and a specific 'mousy' or 'musty' odor the skin or urine of these patients emit.
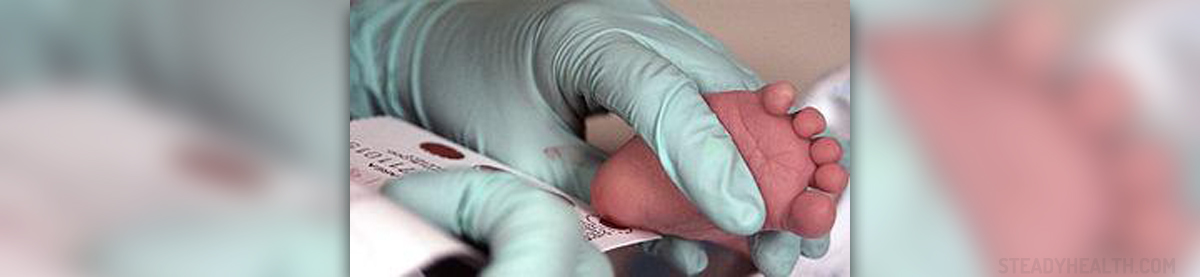
Fortunately, phenylketonuria is easily confirmed with a simple blood test. The test is nowadays performed in the form of screening, as soon as the baby is born. This way the condition can be detected on time. Once the condition is confirmed, the build-up of phenylalanine and associated damage are prevented without difficulty.
Is Phenylketonuria Treatable and what are the Options?
In spite of being a genetic disorder, phenylketonuria may be treated. The treatment comprises a specially planned diet containing no or small amounts of phenylalanine. Patients must stick to their diet, especially growing and developing children. In order to achieve the goal of avoiding the harmful amino acid, children many times require supervision. Consultations with pediatricians and dieticians are frequent. It is estimated that patients who follow their diets into adulthood actually maintain desirable physical and mental health. Additional consultations are necessary once the woman suffering from phenylketonuria decides to get pregnant and during her pregnancy.
Infants suffering from phenylketonuria are not breastfed. Instead they are given a special infant formula Lofenalac. The formula is also good for people of all ages suffering from this condition since it represents an excellent proteins source low in phenylalanine. It contains sufficient amount of other essential amino acids our body requires.
Parents are supposed to keep record on the amount of phenylalanine their child takes on a daily basis. Even once the child enters adolescence, he/she should continue with the diet and remain disciplined for the rest of his/her life, if possible.
Doctors perform frequent blood tests and monitor the level of the amino acid in the blood. This gives insight in whether the diet is efficient enough or needs to be modified.
The best thing is to avoid all high-protein foods. This is why milk and practically all dairy products, eggs, nuts, soybeans, beans, peas, chicken, steak and other beef products, fish, and chocolate are not recommended at all. Furthermore, many artificial sweeteners contain aspartame, a substance that releases phenylalanine once ingested. Aspartame is found in diet sodas and may be also an ingredient of certain medications, all of which are never supposed to be taken. Finally, adults suffering from phenylketonuria should additionally abstain from pasta and rice, bread, cookies and certain fruits and vegetables.
Today certain number of patients is apart from diet prescribed the drug sapropterin. The medication is approved by FDA. The safety and efficacy of the drug are not completely clear yet but they will eventually be thanks to ongoing studies.
Certain supplements may be of additional help. For example, fish oil may replace the long chain fatty acids and boost neurological development. Iron or carnitine may also be recommended.
The prognosis of phenylketonuria is excellent if the condition is confirmed right at birth or shortly after. Only if the disease is misdiagnosed, progressive build-up of phenylalanine affects the central nervous system and triggers a variety of mental and neurological issues. All the infants in whom the condition is not treated from their birth develop mental retardation by the end of the first year of life.
Finally, pregnant women suffering from phenylketonuria are due to pay close attention to their diet. If they choose foods rich in the detrimental amino acid, the acid may pass through the placenta and accumulate in the blood of the unborn baby. So, even though the baby may not have inherited the defective gene, it's brain may get damaged by the excess of amino acid coming from the mother's blood.
Your thoughts on this
Loading...