History of Sickle-cell Disease (SCD)
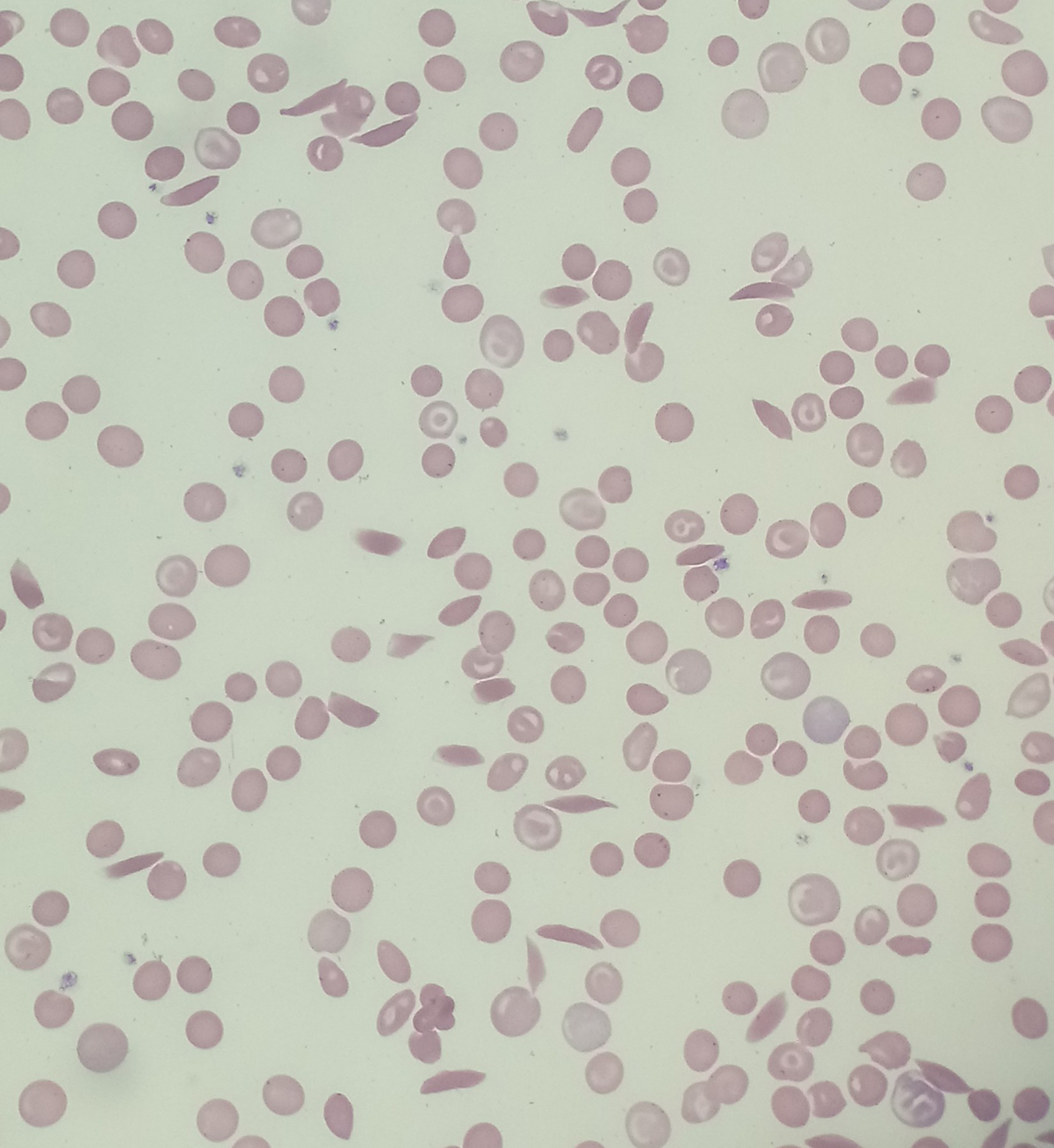
Sickle-cell disease is a genetic blood disorder affecting the shape of red blood cells, making them more susceptible to damage and reducing their lifespan. As a result, the amount of oxygen is not distributed in sufficient amounts to all the body parts. Additionally, the affected red blood cells tend to gather in clusters and cause a variety of health problems and complications. Due to abnormal appearance of erythrocytes and associated health problems, patients suffering from sickle-cell disease have shortened life expectancy. Men in general live up to 42 while women reach 48 years of age. Still, lifespan is individual and may be longer.
The symptoms and signs of sickle-cell disease develop during childhood. The condition is frequent among people living in tropical and sub-tropical regions, especially those where malaria is an endemic disease.
Sickle-cell disease is a broad term used for several conditions. One of these is sickle-cell anemia. Others include sickle-hemoglobin C disease, sickle beta-plus-thalassemia and sickle beta-zero-thalassmia. All of them are characterized by the presence of an abnormal gene, the one in charge with production of hemoglobin of normal appearance and function.
In the United States sickle-cell disease affects 1 out of 5,000 people who are mostly of Sub-Saharan African descent. As a matter of fact 1 out of 500 African-American children inherit the defective gene and develop this disease.
Sickle-cell Conditions are Inherited from Parents
The condition develops as a result of a point mutation in the β-globin chain of hemoglobin. This gene is localized on the short arm of the 11th chromosome.
The defect in the mentioned gene is blamed for the loss of erythrocyte elasticity. Under normal circumstances, red blood cells are elastic which enables them to pass through capillaries without being damaged. In patients suffering from sickle-cell disease red blood cells become sickle in shape and have significantly reduced elasticity, meaning they get easily damaged once they come in contact with capillaries. Vessel occlusion and ischemia are two more problems red blood cells shaped in such a way are responsible for.
Scientists believe that sickle-cell gene mutation arose spontaneously in certain geographic areas. People who carry the gene may be heterozygous or homozygous for HgbS. In the first case the person may produce normal hemoglobin and it accounts for more than 50% of all produced hemoglobin. In the second case, when a person is homozygous for HgbS practically the entire hemoglobin is abnormal and all the red blood cells are misshapen.
The condition is inherited in an autosomal recessive pattern. In case both parents have the defective gene the child has 25% chance to inherit the diseases. Furthermore, if one parent is suffering from sickle-cell disease and the other has sickle-cell trait the child will in 50% inherit the disease and there is 50% chance that the trait is going to be inherited. Potential Complications
There is a range of complications such patients may eventually face with. For example, one is prone to overwhelming post- (auto) splenectomy infections. The susceptibility to infections may be dealt with penicillin prophylaxis and adequate/timely vaccinations. Another common complication of sickle-cell disease is stroke. It develops due to progressive narrowing of blood vessels in the brain which eventually results in a complete decrease of blood supply to certain portions of the organ. Even though stroke may be symptomatic, in sickle-cell disease patients it is more likely to be silent.
Furthermore, these patients are prone to cholelithiasis, osteomyelitis and avascular/aseptic bone necrosis. The immune system becomes weaker due to hyposplenism. The eyes may be affected as well, especially with different types of retinopathy, vitreous hemorrhage and retinal detachment. One may develop leg ulcers as well. In men there is higher risk of priapism and infarction of the penis while in women there may be intrauterine growth retardation, spontaneous abortion and preeclampsia, all associated with pregnancy.
Patients with sickle-cell disease suffer chronic pain and many of them develop opoid tolerance, a psychological response to the therapeutic use of opiates. Also, these patients are at risk of pulmonary hypertension, acute papillary necrosis in the kidneys and chronic renal failure.
Treatment Options
Children who have been diagnosed sickle-cell disease are closely monitored by their pediatricians and hematologists. They are administered folic acid on a daily basis and also receive penicillin prophylactically until the age of 5. This is a preventative measure against frequent infections.
Vaso-occlusive crises are very painful and one of the major health issues for sickle-cell disease patients. The pain is alleviated with analgesics such as opoids. Milder crisis can be dealt with NSAIDs. Finally, severe pain is brought under control with intravenous opoids with the assistance of patient-controlled analgesia (PCA) devices.
Acute chest crisis is treated in the same manner as vaso-occlusive crisis with additional antibiotics and oxygen supply. Blood transfusions and exchange transfusion may be necessary as well.
Hydroxyurea is the only drug that has been proven efficient in these patients and may be prescribed if necessary. The drug decreases the severity of attacks. The effects of long-term use of hydroxyurea are still being investigated.
Finally, one more treatment approach is bone marrow transplant. It is generally recommended in children.
Your thoughts on this
Loading...