Definition
Marfan syndrome belongs to genetic disorders predominantly affecting connective tissue or different organs and organ systems. Individuals suffering from Marfan syndrome are unusually tall with long upper and lower extremities and long, thin fingers.
Marfan syndrome is inherited in an autosomal dominant fashion. The defective gene responsible for the syndrome has been identified and named FBN1 gene. A person needs to inherit only one defective gene from either mother or father in order to develop Marfan syndrome.
The very syndrome is characterized by a variety of structural and functional changes, all of which may be mild or sometimes severe in nature. In the majority of cases there are defects on the heart valves and aorta. Furthermore, there is damage to the lungs, eyes and membrane surrounding the spinal cord. Skeletal deformities are also typical for people suffering from Marfan syndrome.
Symptoms and Signs
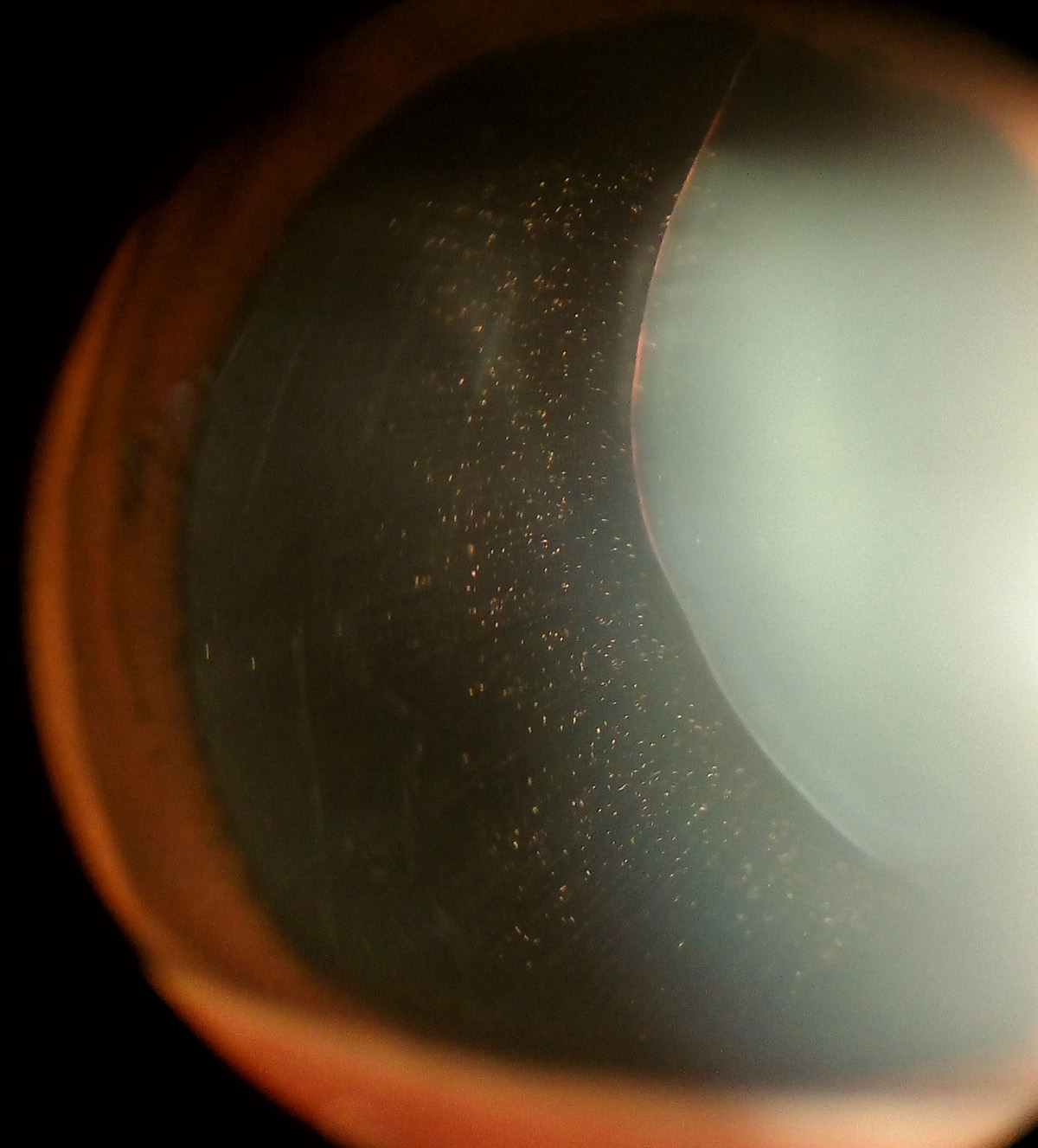
Apart from being responsible for many numerous structural and functional changes, Marfan syndrome is easily confirmed if a person has long extremities and suffers from dislocated lenses and the aortic root dilation.
As far as skeletal deformities are concerned, patients with Marfan syndrome are abnormally high (have long bones), their extremities are slender and both fingers and toes are unusually thin and long which is medically known as arachnodactyly. Patients are additionally prone to scoliosis and kyphosis(spinal abnormality) and pectus excavatum/pectus carinatum (abnormalities of the breast bone and rib cage).
Oral skeletal deformities occur in the form of a high palate, small jaws and malocclusion. Patients may have flat feet, incredibly flexible joints and suffer from stretch marks on the skin. Certain number of patients also develop osteoarthritis.
Marfan syndrome is a cause of lens dislocation and is associated with several more vision problems. Namely, such patients are prone to nearsightedness and astigmatism. Since connective tissue is normally affected by the syndrome it is not unusual if certain number of patients eventually develop retinal detachment.
As for cardiovascular system, Marfan syndrome may cause broad spectrum of illnesses including angina, circulatory issues, defects or damage to heart valves (especially prolapse of the mitral or aortic valves), aortic aneurysm, aortic dissection etc. Many such conditions require surgical correction.
Women suffering from this syndrome are especially susceptible to certain cardiovascular complications such as aortic dissection. Because of that they must be thoroughly monitored throughout their pregnancy.
Respiratory issues affecting these patients include proneness to spontaneous pneumotorax, sleep apnea and idiopathic obstructive lung disease.
Dural ectasia is a common complication of the weakening of the connective tissue of the dural sac and it is blamed for a range of neurological symptoms and signs some of which are recurrent or even persistent. These include back pain (usually lower back pain), leg pain, pain in the abdominal area, headaches etc.
Finally, additionally health problems these patients may face with are GERD, degenerative disc disorder, deviated septum, early cataract or glaucoma, osteopenia, osteoarthritis, emphysema, hernia and temporomandibular joint disorder.
Management
Since Marfan syndrome is a condition that develops as a consequence of gene defects, it simply cannot be cured. However, most conditions the syndrome brings on are treatable and can be dealt with successfully.
Because of the mentioned, doctors typically focus on medical issues patients are currently having problems with. Some conditions are treated conservatively, while others require surgery. It is also possible to prevent certain conditions from occurring. For instance, progression of aortic dilation is decelerated with some medications.
Having regular check-ups is a must. This way doctors have insight in progression of ongoing conditions and the onset of new health problems. This particularly refers to cardiovascular examination when most heart diseases can be diagnosed on time and treated accordingly. Conservative treatment basically comprises medications such as beta blockers, ACE inhibitors while surgery is reserved for patients in whom aortic dilation and damage to heart valves is severe and has detrimental effects on the entire body additionally putting patients at risk of more severe complications. Surgery is also performed in case of acute aortic dissection and acute aortic rupture.
Fortunately, skeletal manifestation of the syndrome are not that serious and are easily brought under control with various drugs like pain killers, muscle relaxants and physical therapy. Surgical corrections are only indicated in more complex deformities.
All in all, since Marfan syndrome is passed on to offspring, by performing genetic testing of parents, the doctor may estimate the chances of transmission of the defective gene and leave parents to chose whether they will conceive a child or decide not to have children.
Statistical Data
It is estimated that Marfan syndrome affects both genders almost equally. The genetic defects are also equally present in all ethnic groups. According to certain data the syndrome occurs in 1 out of 3,000-5,000 individuals.
If one parent suffers from Marfan syndrome, there is 50% chance that the child will develop the same condition.
Even though in the majority of cases there are other family members suffering from Marfan syndrome in around 15-30% of all cases the condition develops as a result of new, spontaneous mutations.
Your thoughts on this
Loading...