Definition of Idiopathic Pulmonary Fibrosis (IPF)
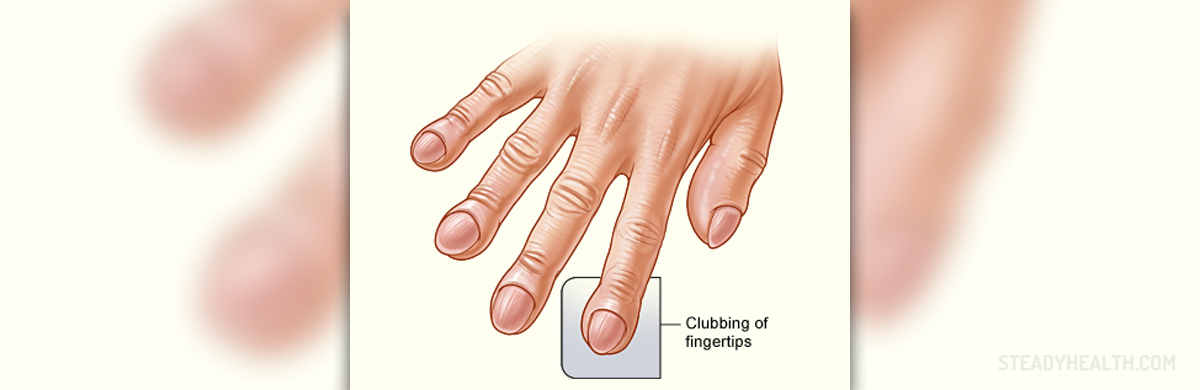
Idiopathic pulmonary fibrosis is a continuous lung disorder characterized by the scarring of the tissue of the lungs. Despite many clinical studies that have been conducted to identify the root cause of the disorder, it still remains unknown. Individuals who are experiencing a dry cough for longer than a month, coupled with difficulties during breathing, should consult with a medical care provider and have themselves checked for IPF, as those are its usual symptoms. The process that takes place in IPF is the inflammation of the alveoli caused by excess amount of collagen, which is usually induced by smoking or various kinds of autoimmune disorders. In and of itself, the idiopathic pulmonary fibrosis is a type of pneumonia with the highest prevalence among the middle aged and across both sexes. As far as living with the disease goes, different people have different experiences, but for the most part few years pass before the disorder becomes severely debilitating. The most common problems associated with idiopathic pulmonary fibrosis that most patients deal with are dry cough, deforming of the fingers and difficulties breathing. In addition, there are many medical conditions that can contribute to the development of IPF, such as arthritis and various respiratory problems.
Diagnosis of Idiopathic Pulmonary Fibrosis
As is the case with many medical conditions, the symptoms of idiopathic pulmonary fibrosis are comorbid with numerous other problems, some respiratory and others not. The first challenge that medical professionals are faced with is identifying the underlying cause of the symptoms. For instance, in some cases it is complicated to differentiate between the signs of arthritis, being exposed to toxic substances and various types of neurological disorders, such as sclerosis and the symptoms of IPF. Further, diagnosing idiopathic pulmonary fibrosis usually entails radiological tests as well as biopsies of the lungs. The biopsies are more often than not avoided if the doctors can agree that the symptoms are in fact due to IPF. One of the reasons why biopsies are avoided is because a relatively large sample of the tissue is necessary in order for the pathologists to detect changes and such large samples must be obtained surgically. When it comes to scanning, x - rays are widely used to diagnose IPF, usually by searching for cysts, as well as lower volume of the lungs. The more advanced the stage of IPF, the more evident are the changes that could be detected via x – rays. There are instances when CT scans are used as well, mainly to look for scarring in the lungs. Further, a test that is also used to check for IPF is spirometry, which also reveals the decrease in the volume of the lungs as well as their reduced flexibility. In general, if an individual who is exhibiting the signs of IPF hasn’t been taking any medications that produce IPF like symptoms, hasn’t been suffering from pulmonary disorders and hasn’t been in contact with toxic substances, there is a strong possibility that such a patient is affected by IPF. Further, if the tests show impairment in oxygen transfer or the volume of the lungs coupled with symptoms of IPF, it can also be concluded that the underlying illness is in fact IPF. Finally, if all other illnesses with similar symptoms can be ruled out by the process of deductive reasoning clinicians will diagnose a patient with idiopathic pulmonary fibrosis. In individuals who are over 50 years old, with previous respiratory problems and the symptoms for more than 3 months, the IPF is relatively easily diagnosed.
Treatment and Prognosis for Idiopathic Pulmonary Fibrosis
At the moment, there is no treatment for idiopathic pulmonary fibrosis and every individual case is dealt with separately. One of the factors contributing to the lack of adequate treatment is a deficit in research that deals with IPF therapy options. Until recently, there was a lot of confusion with the classification of patients and their symptoms in clinical studies so the results are not valid. There have been many experiments that looked at the effects of various types of medications on IPF, but more research is needed in order to come up with the right kind of therapy. As far as prognosis goes, lots of individuals have to have oxygen tanks on them at all times and in some cases a lung transplant, but those are rare. In the UK, about 50% of those diagnosed with idiopathic pulmonary fibrosis die within the first 3 years, while overall about 75% of individuals diagnosed with IPF die within 5 years. In the US, the number of people who die from IPF every year is about the same as the number of people who die from breast cancer. Finally, in recent years the IPF has become at least 5 times more prevalent than in the previous decades, probably because of better defined diagnostic criteria.
Your thoughts on this
Loading...