Huntington's disease is a genetic disorder characterized by a palette of symptoms and signs caused by damage to the brain. The first symptoms and signs of Huntington's disease occur when patients enter the fourth or the fifth decade of their lives. They experience a range of health issues including a gradual loss of control of movement, memory and mental ability.
Even though the condition is practically incurable, there are treatments available that can bring some symptoms and signs of the disease under control and improve patients' quality of life.
Huntington's Disease - Who is at Risk?
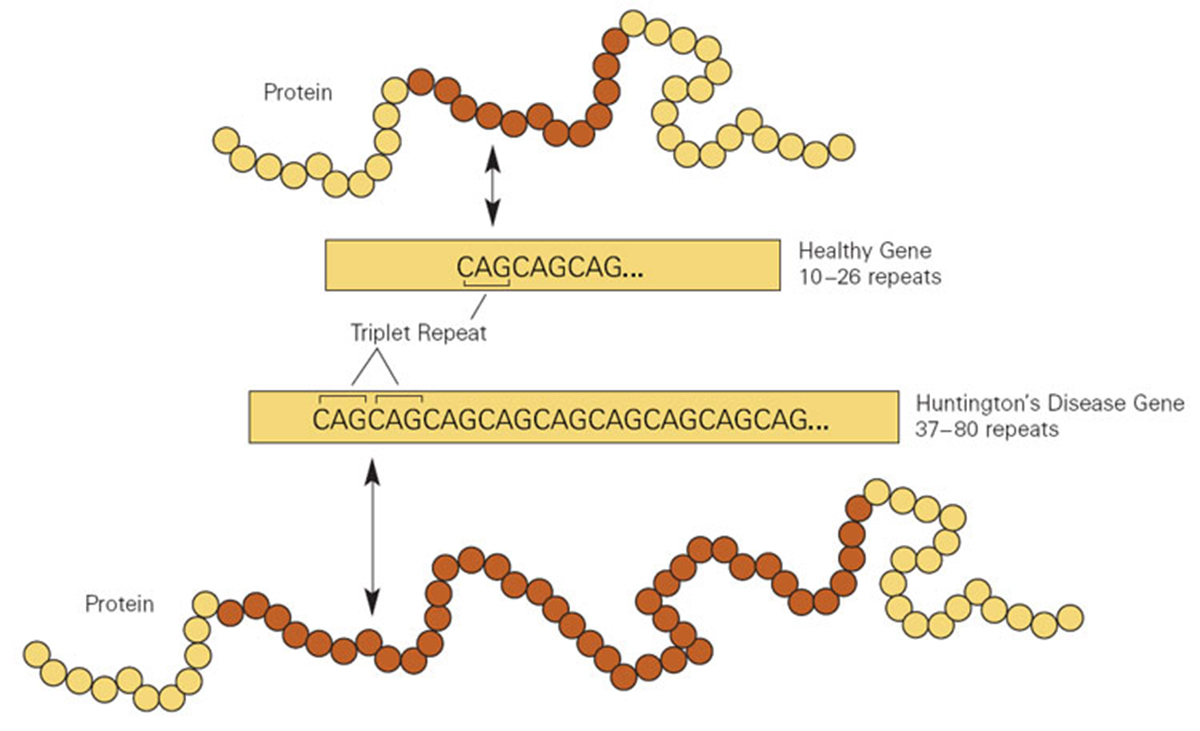
Since Huntington's disease is a genetic disorder it is clear that the condition cannot be acquired but it is only inherited from parents. The disease runs in some families and there may even be a family history when Huntington's disease affects several family members. On the other hand, the disease may have been present in previous generations but younger members of the family are not aware of that fact so they cannot for sure say whether there have been cases of the disease running in the family.
This genetic disease is inherited in an autosomal dominant pattern. The chances of receiving a faulty gene if one parent is suffering from Huntington's disease are 50%.
Huntington's Disease - Symptoms and Signs
Huntington's disease is blamed for the loss of numerous brain functions. The first symptoms and signs occur in the middle ages. These patients may first experience changes in personality or sometimes mood changes. These can be easily neglected but when patients eventually start to experience problems with memory and abnormal movements they do turn to their health care providers.
It may sound amazing but personality changes may occur as much as ten days before abnormal movements start.
Once the symptoms show up, they progress unstoppably and become worse in time. The degree of involuntary movement is different among patients and the lucky ones end up with only a few involuntary movements.
Lethal outcome is reported to occur approximately 15-20 years after the onset of first symptoms. Death is associated either with a general decline in health or for instance, some may choke on food due to swallowing difficulty.
Huntington's Disease - Eye Problems
As far as patients suffering from Huntington's disease are concerned, there are no serious problems with their eyes. However, some of them may experience rapid eye movement, especially on looking up or down. These patients additionally have problems when looking sideways. This is why they move their head in the desired direction in order to achieve full lateral vision.
- While HTT expansion repeat transgenics have been shown to exhibit retinal photoreceptor degeneration, these models were hampered by artificially long expansions that may cause ‘off effects’. We wanted to evaluate retinal function and structure in an HTT model that more closely resembles the human disease.
- Transgenic knock-in Huntington’s rats containing the human HTT gene with a 60-75 expansion repeat were used. Both Wild type (WT) and mutant pairs from the same litters were evaluated, and at different ages.
- Following at least 12 hours of dark-adaptation, we measured electroretinograms (ERGs) in pairs of HTT rats and their WT littermates, using the ISCEV protocol and an additional paradigm we developed to expose changes in presumed horizontal cell function.
- No differences were seen in the ERG a- and b-waves of the HTT and normal rats. However, HTT rats showed reduced oscillatory potential amplitudes and disinhibition of the photopic response.
- Immunohistochemistry showed HTT accumulation in the horizontal cells and suggested a loss of ChAT (+)-amacrine cells.
Your thoughts on this
Loading...